Search
164 results found
- MERTK missense variants in three patients with retinitis pigmentosa
Federica E. Poli, Imran H. Yusuf, Penny Clouston, Morag Shanks, Jennifer Whitfield, Peter Charbel Issa, Robert E. MacLaren | Ophthalmic Genetics | 29 Aug 2022 | 44 (1) | pgs. 74-82 | doi.org/10.1080/13816810.2022.2113541 Abstract Background MERTK (MER proto-oncogene, tyrosine kinase) is a transmembrane protein essential in regulating photoreceptor outer segment phagocytosis. Biallelic mutations in MERTK cause retinal degeneration. Here we present the retinal phenotype of three patients with missense variants in MERTK . Materials and methods All patients underwent a full clinical examination, fundus photography, short-wavelength fundus autofluorescence and optical coherence tomography imaging. Two patients also underwent Goldmann visual field testing and electroretinography was undertaken for the third patient. Molecular genetic testing was undertaken using next generation or whole-exome sequencing with all variants confirmed by Sanger sequencing. Results The first patient was a 29-year-old female heterozygous for a missense variant (c.1133C>T, p.Thr378 Met) and a nonsense variant (c.1744_1751delinsT, p.Ile582Ter) in MERTK . The second patient was a 26-year-old male homozygous for a c.2163T>A, p.His721Gln variant in MERTK . The third patient was an 11-year-old female heterozygous for a deletion of exons 5–19 and a missense variant (c.1866 G>C, p.Lys622Asn) in MERTK . Reduced night vision was the initial symptom in all patients. Fundoscopy revealed typical signs of retinitis pigmentosa (RP) with early-onset macular atrophy. All three MERTK missense variants affect highly conserved residues within functional domains, have low population frequencies and are predicted to be pathogenic in silico . Conclusions We report three missense variants in MERTK and present the associated phenotypic data, which are supportive of non-syndromic RP. MERTK is a promising candidate for viral-mediated gene replacement therapy. Moreover, one variant represents a single nucleotide transition, which is theoretically targetable with CRISPR-Cas9 base-editing. Introduction Retinitis pigmentosa (RP) is a set of clinically and genetically heterogenous inherited retinal dystrophies characterised by progressive primary rod photoreceptor degeneration with concurrent, or later degeneration of cones ( Citation1 ). It typically manifests with difficulties in dark adaptation and night vision, followed by progressive peripheral visual field loss, with subsequent loss of central vision. A vast number of heterogenous genetic defects have been implicated in the pathogenesis of non-syndromic RP ( Citation2 , Citation3 ). The MER proto-oncogene, tyrosine kinase ( MERTK ) gene encodes a transmembrane protein expressed in the retinal pigment epithelium (RPE), which plays a critical role in photoreceptor homeostasis by regulation of phagocytosis of shed photoreceptor outer segment discs ( Figure 1 , panel A) ( Citation4 , Citation5 ). Numerous mutations in MERTK have been identified as pathogenic for retinal dystrophies ( Citation6 , Citation7 ). MERTK mutations cause a rod-cone dystrophy with early macular atrophy, with RP being the most common retinal phenotype, although cases of Leber congenital amaurosis have been reported ( Citation7 ). Figure 1. Panel A: structure of the MERTK transmembrane protein. The extracellular portion includes two immunoglobulin-like (Ig-like) domains (green) and two fibronectin type III (FN-III) domains (blue). The intracellular region contains a highly conserved kinase domain (yellow). The location of the Three mutations discussed are indicated by red arrows. The respective amino acid residues corresponding to the domains affected are indicated below the protein schematic. Panel B: Conservation across species of the amino acid residue subject to mutation in the c.1133c>t, p.Thr378 met variant (B1), the c.2163t>a, p.His721gln variant (B2) and the c.1866G>C, p.Lys622Asn variant (B3). Panel C: Pedigrees for Case 1 (C1) and Case 2 (C2). Pedigree not available for Case 3. Note the pedigrees are based on clinical presentation and segregation has not been possible. Click here to read entire paper References Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–809. doi:10.1016/S0140-6736(06)69740-7. RetNet: summaries of genes and loci causing retinal diseases. 2021. https://sph.uth.edu/retnet/sum-dis.htm#A-genes(open in a new window) . Verbakel SK, van Huet RA, Boon CJ, den Hollander AI, Collin RW, Klaver CC, Hoyng CB, Roepman R, Klevering BJ. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86. Feng W, Yasumura D, Matthes MT, LaVail MM, Vollrath D. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem. 2002;277(19):17016–22. doi:10.1074/jbc.M107876200. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11):a009076. doi:10.1101/cshperspect.a009076. Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26(3):270–71. doi:10.1038/81555. Audo I, Mohand‐said S, Boulanger‐scemama E, Zanlonghi X, Condroyer C, Démontant V, Boyard F, Antonio A, Méjécase C, El Shamieh S, Sahel JA. MERTK mutation update in inherited retinal diseases. Hum Mutat. 2018;39(7):887–913. doi:10.1002/humu.23431. Ellingford JM, Barton S, Bhaskar S, O’Sullivan J, Williams SG, Lamb JA, Panda B, Sergouniotis PI, Gillespie RL, Daiger SP, et al. Molecular findings from 537 individuals with inherited retinal disease. J Med Genet. 2016;53(11):761–67. doi:10.1136/jmedgenet-2016-103837. Takahashi VK, Xu CL, Takiuti JT, Apatoff MBL, Duong JK, Mahajan VB, Tsang SH. Comparison of structural progression between ciliopathy and non-ciliopathy associated with autosomal recessive retinitis pigmentosa. Orphanet J Rare Dis. 2019;14(1):1–9. doi:10.1186/s13023-019-1163-9. Colombo L, Maltese PE, Castori M, El Shamieh S, Zeitz C, Audo I, Zulian A, Marinelli C, Benedetti S, Costantini A, et al. Molecular epidemiology in 591 Italian probands with nonsyndromic retinitis pigmentosa and usher syndrome. Invest Ophthalmol Vis Sci. 2021;62(2):13. doi:10.1167/iovs.62.2.13. Shanks ME, Downes SM, Copley RR, Lise S, Broxholme J, Hudspith KA, Kwasniewska A, Davies WI, Hankins MW, Packham ER, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013;21(3):274–80. doi:10.1038/ejhg.2012.172. D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9(4):645–51. doi:10.1093/hmg/9.4.645. Nandrot E, Dufour EM, Provost AC, Péquignot MO, Bonnel S, Gogat K, Marchant D, Rouillac C, Sépulchre de Condé B, Bihoreau M-T, et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mechanisms of physiological cell adhesion and apoptosis. Neurobiol Dis. 2000;7(6):586–99. doi:10.1006/nbdi.2000.0328. Mullen RJ, LaVail MM. Inherited retinal dystrophy: primary defect in pigment epithelium determined with experimental rat chimeras. Science. 1976;192(4241):799–801. doi:10.1126/science.1265483. Dowling JE, Sidman RL. Inherited retinal dystrophy in the rat. J Cell Biol. 1962;14(1):73–109. doi:10.1083/jcb.14.1.73. Bok D, Hall MO. The role of the pigment epithelium in the etiology of inherited retinal dystrophy in the rat. J Cell Biol. 1971;49(3):664–82. doi:10.1083/jcb.49.3.664. Jonsson F, Burstedt M, Kellgren TG, Golovleva I Non-Homologous recombination between Alu and LINE-1 repeats results in a 91 kb deletion in MERTK causing severe retinitis pigmentosa. Mol Vis. 2018;24:667. Liu S, Bi JG, Hu Y, Tang D, Li B, Zhu P, Peng W, Du D, He H, Zeng J, et al. Targeted next generation sequencing identified novel loss-of-function mutations in MERTK gene in Chinese patients with retinitis pigmentosa. Mol Genet Genomic Med. 2019;7(4):e00577. doi:10.1002/mgg3.577. Jespersgaard C, Bertelsen M, Arif F, Gellert-Kristensen HG, Fang M, Jensen H, Rosenberg T, Tümer Z, Møller LB, Brøndum-Nielsen K, et al. Bi-allelic pathogenic variations in MERTK including deletions are associated with an early onset progressive form of retinitis pigmentosa. Genes. 2020;11(12):1517. doi:10.3390/genes11121517. Birtel J, Eisenberger T, Gliem M, Müller PL, Herrmann P, Betz C, Zahnleiter D, Neuhaus C, Lenzner S, Holz FG, et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci Rep. 2018;8(1):1–11. doi:10.1038/s41598-018-22096-0. Issa PC, Bolz HJ, Ebermann I, Domeier E, Holz FG, Scholl HP. Characterisation of severe rod–cone dystrophy in a consanguineous family with a splice site mutation in the MERTK gene. Br J Ophthalmol. 2009;93(7):920–25. doi:10.1136/bjo.2008.147397. Gliem M, Müller PL, Birtel J, Herrmann P, McGuinness MB, Holz FG, Charbel Issa P. Quantitative fundus autofluorescence and genetic associations in macular, cone, and cone–rod dystrophies. Ophthalmol Retina. 2020;4(7):737–49. doi:10.1016/j.oret.2020.02.009. Strick DJ, Vollrath D. Focus on molecules: MERTK. Exp Eye Res. 2010;91(6):786. doi:10.1016/j.exer.2010.05.006. Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, Bieg A, Gliem M, Issa PC, Holz FG, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PloS One. 2013;8(11):e78496. doi:10.1371/journal.pone.0078496. Cehajic-Kapetanovic J, Xue K, de la Camara, A Nanda, Martinez-Fernandez C, Nanda LJ, Davies A, Wood, LJ, Fischer, MD, Aylward, JW, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. 2020;26(3):354–59. doi:10.1038/s41591-020-0763-1. Xue K, Jolly JK, Barnard AR, Rudenko A, Salvetti AP, Patrício MI, Edwards, TL, Groppe, M, Orlans, HO, Tolmachova, T, et al. Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med. 2018;24(10):1507–12. doi:10.1038/s41591-018-0185-5. Russell S, Bennett J, Wellman JA, Chung DC, Yu Z, Tillman A, Wittes, J, Pappas, J, Elci, O, McCague, S, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRpe65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–60. doi:10.1016/S0140-6736(17)31868-8. Askou AL, Jakobsen TS, Corydon TJ Retinal gene therapy: an eye-opener of the 21st century. Gene Ther. 2021;28(5):209–16. doi:10.1038/s41434-020-0168-2. Tschernutter M, Schlichtenbrede FC, Howe S, Balaggan KS, Munro PM, Bainbridge J, Thrasher, AJ, Smith, AJ, Ali, RR Long-Term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther. 2005;12(8):694–701. doi:10.1038/sj.gt.3302460. Smith AJ, Schlichtenbrede FC, Tschernutter M, Bainbridge JW, Thrasher AJ, Ali RR AAV-Mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8(2):188–95. doi:10.1016/S1525-0016(03)00144-8. Vollrath D, Feng W, Duncan JL, Yasumura D, D’Cruz PM, Chappelow A, Matthes, MT, Kay, MA, LaVail, MM Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Nat Acad Sci. 2001;98(22):12584–89. doi:10.1073/pnas.221364198. Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai H, Hou, R, Deng, W-T, Boye, SL, Almaghamsi, A, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. 2016;135(3):327–43. doi:10.1007/s00439-016-1637-y. Li T, Adamian M, Roof DJ, Berson EL, Dryja TP, Roessler BJ, Davidson, BL In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest Ophthalmol Vis Sci. 1994;35(5):2543–49. Bennett J, Wilson J, Sun D, Forbes B, Maguire A Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci. 1994;35(5):2535–42. Dong J, Fan P, Frizzell RA Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther. 1996;7(17):2101–12. doi:10.1089/hum.1996.7.17-2101. Rees HA, Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–88. doi:10.1038/s41576-018-0059-1. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–24. doi:10.1038/nature17946. Cox DB, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang, F. RNA editing with CRISPR-Cas13. Science. 2017;358(6366):1019–27. doi:10.1126/science.aaq0180. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu, DR Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71. doi:10.1038/nature24644. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen, PJ, Wilson, C, Newby, G A, Raguram, A., et al. Search-And-Replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57. doi:10.1038/s41586-019-1711-4.
- Recent Advances of Stem Cell Therapy for Retinitis Pigmentosa
He, Y., Zhang, Y., Liu, X., Ghazaryan, E., Li, Y., Xie, J., & Su, G. (2014). Recent advances of stem cell therapy for retinitis pigmentosa. International journal of molecular sciences , 15 (8), 14456–14474. https://doi.org/10.3390/ijms150814456 Abstract Retinitis pigmentosa (RP) is a group of inherited retinal disorders characterized by progressive loss of photoreceptors and eventually leads to retina degeneration and atrophy. Until now, the exact pathogenesis and etiology of this disease has not been clear, and many approaches for RP therapies have been carried out in animals and in clinical trials. In recent years, stem cell transplantation-based attempts made some progress, especially the transplantation of bone marrow-derived mesenchymal stem cells (BMSCs). This review will provide an overview of stem cell-based treatment of RP and its main problems, to provide evidence for the safety and feasibility for further clinical treatment. Introduction Retinitis pigmentosa (RP) is a group of inherited retinal disorders characterized by progressive loss of photoreceptors and eventually leads to retina degeneration and atrophy. New approaches for RP therapies include: cell transplantation therapy, gene therapy, cytokine therapy, nutrition therapy, and hyperbaric oxygen therapy. Present therapies for RP are restricted in their efficacy or safety, for example: maintenance of long-term efficacy using a single injection of cytokines is difficult, but there is a risk of infection after repeated intra-vitreous injections in cytokine therapy. Gene therapy has been shown to improve visual function in inherited retinal disease. RP is a hereditary cause of blindness, which has four main modes of inheritance: Autosomal dominant RP (ADRP), autosomal recessive RP (ARRP), X-linked RP (XLRP) and dihybrid inheritance, also mitochondrial genetic and non-genetic forms. Thomas and colleagues injected rAAV2-VMD2-hMERTK vector into the sub-retinal space of RCS rats and SD rats; it showed improvement of visual function in RCS, they also performed a series of safety studies in normal SD rats, and demonstrated that no local or systemic toxicity was detected after either dose of vector delivery and no indication of vector spread outside the treated eye. This group also prompted a phase I clinical trial in which rAAV2-VMD2-hMERTK vector were injected into the sub-retinal space of patients with retinal disease due to MERTK mutations. To continue reading the paper, click here .
- Retinitis pigmentosa with or without skeletal abnormalities due to homozygous mutations in the CWC27 gene: A case report
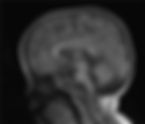
Qi, Yang-Fan MDa; Ma, Xiaoping MDb; Lin, Shuang-Zhu PhDc; Wang, Wan-Qi MDa; Li, Jia-Yi MDa; Chen, Qian-Dui MDa; Liu, Li PhDb, | Medicine | December 22, 2023 | 102(51) | p e36357 | DOI: 10.1097/MD.0000000000036357 Abstract Rational Retinitis pigmentosa with or without skeletal abnormalities (RPSKA) is an autosomal recessive disorder caused by mutations in the CWC27 gene. Skeletal dysplasia and non-syndromic retinitis pigmentosa are typical manifestations, and most patients present with retinopathy such as retinitis pigmentosa and limited visual field. Its clinical manifestations are complex and diverse, often involving multiple systems. Examples include short finger deformities, peculiar facial features, short stature, and neurodevelopmental abnormalities, and it is easy to misdiagnose clinically, and early diagnosis is crucial for prognosis. Patient concerns A 2-year and 2-month-old female child was admitted to the hospital due to “unsteady walking alone and slow reaction for more than half a year.” After admission, the child was found to have delayed motor development, accompanied by special face, abnormal physical examination of the nervous system, cranial MRI Dandy-Walker malformation, considering developmental delay. Diagnoses Whole exome sequencing of the family line revealed the presence of a c.617(exon7)C>A pure mutation in the CWC27 gene in the affected child (this locus has been reported in the clinical literature); the final diagnosis is RPSKA. Interventions Unfortunately, there is no specific drug for the disease; we give children rehabilitation training treatment. Outcomes During follow-up process we found that children’s condition is better than before. Lessons subsections as per style We reported a case of RPSKA caused by mutations in the CWC27 gene. This study adds to our understanding of the clinical phenotype of TBL1XR1 mutations and provides a realistic and reliable basis for clinicians. Introduction The CWC27 gene (OMIM: 617170), located on chromosome 5q12.3, is a splice-like cyclophilic peptidyl-prolyl cis-trans isomerase[ 1 ]; Phillips et al[ 2 ] first reported children with the gene mutation in 1981. Inheritance is autosomal recessive, and common variants include homozygous and complex heterozygous variants. Retinitis pigmentosa with or without skeletal anomalies (RPSKA) (OMIM: 250410) is an autosomal recessive genetic disease caused by mutations in the CWC27 gene. The disease can involve multiple systems, with skeletal dysplasia and retinopathy as the main manifestations, accompanied by developmental abnormalities. The characteristic retinopathy is manifested as retinitis pigmentosa, restricted visual field, ptosis of eyelid fissure, and thinning of retinal blood vessels.[ 3 , 4 ] We report a case of a homozygous variant of the CWC27 gene in which onset of stunting was the main manifestation. Our report enriches the understanding of the clinical phenotype of CWC27 mutations and provides a realistic and reliable basis for clinicians. Case presentation A 2-year and 2-month-old female child suffered from gross motor developmental delay after birth. She could stand upright at 2 months, turn over at 4 months, sit at 6 months, and unconsciously utter “baba, mama” at 1 year and 1 month. Sound, crawling at 1 year and 4 months, walking alone at 1 year and 8 months, still walking unsteadily, slow response, can only consciously pronounce “Dad, Mom.” During the period, the child was 8 months old because she could not climb and was treated in our hospital, and the physical examination found that the muscle tone was high, and there was no significant improvement after giving rehabilitation training guidance, and half a month ago she was treated in our hospital because of “unstable walking alone and slow response,” and was admitted to the hospital after the outpatient clinic with the main complaint of “unstable walking alone and slow response for more than half a year.” Current symptoms: the child is unstable alone, slow response, can only consciously make “dad, mother” sound, no cough and fever, no vomiting and diarrhea, diet and sleep, normal stool. After admission, we asked the children’s birth and growth history in detail; the children were G4P2, 36 + 6 weeks cesarean section, BW 2000 g (−3SD), born with little amniotic fluid, asphyxiation and rescue history, post-natal diagnosis of “respiratory distress, preterm delivery, low birth weight infants, neonatal pneumonia, neonatal hypoglycemia, ventricular, atrial, patent ductus arteriosus, hyperbilirubinemia, renal horseshoe kidney, hearing abnormalities of both ears,” after treatment were better discharged (unknown). The mother had been suffering from “upper respiratory tract infection” for more than 10 days during pregnancy, without special treatment, before birth due to “little amniotic fluid” oxygen for 2 days, and still denied the special situation. After birth, the children’s great movement development lags behind their peers, they can erect at the age of 2 months, turn over at the age of 4 months, sit at the age of 6 months, unconsciously give the sounds of “baba, mama” at the age of 1 year, climb at the age of 1 year, 4 months, walk alone at the age of 1 year, and still walk unstably, have slow reaction, have slow eye contact with people, can express sign language, but can only give the sounds of “Mom and Dad,” are happy to talk to themselves, are happy to open the door, can execute simple instructions, cannot show the inconvenience. Continue reading entire article
- ARL2BP, a protein linked to retinitis pigmentosa, is needed for normal photoreceptor cilia doublets and outer segment structure
Abigail R Moye , Ratnesh Singh , Victoria A Kimler , Tanya L Dilan , Daniella Munezero , Thamaraiselvi Saravanan , Andrew F X Goldberg , Visvanathan Ramamurthy | Molecular Biology of the Cell | 2018 Jul 1 | Vol 29, Iss 13 | pgs. 1590–1598 | doi: 10.1091/mbc.E18-01-0040 Abstract The outer segment (OS) of photoreceptor cells is an elaboration of a primary cilium with organized stacks of membranous disks that contain the proteins needed for phototransduction and vision. Though ciliary formation and function has been well characterized, little is known about the role of cilia in the development of photoreceptor OS. Nevertheless, progress has been made by studying mutations in ciliary proteins, which often result in malformed OSs and lead to blinding diseases. To investigate how ciliary proteins contribute to OS formation, we generated a knockout (KO) mouse model for ARL2BP, a ciliary protein linked to retinitis pigmentosa. The KO mice display an early and progressive reduction in visual response. Before photoreceptor degeneration, we observed disorganization of the photoreceptor OS, with vertically aligned disks and shortened axonemes. Interestingly, ciliary doublet microtubule (MT) structure was also impaired, displaying open B-tubule doublets, paired with loss of singlet MTs. On the basis of results from this study, we conclude that ARL2BP is necessary for photoreceptor ciliary doublet formation and axoneme elongation, which is required for OS morphogenesis and vision. Introduction Photoreceptors are ciliated neurons that absorb photons and convert light into electrical signals. These neurons are compartmentalized with outer and inner segments (OS and IS) bridged by a narrow connecting cilium (CC, corresponds to the ciliary transition zone) with an extended axoneme ( Pearring et al. , 2013 ; Goldberg et al. , 2016 ; May-Simera et al. , 2017 ). The OS contains stacked membranous disks that anchor proteins that participate in phototransduction ( Molday and Moritz, 2015 ). Remarkably, photoreceptors shed 10% of their disks every day ( Young, 1967 ; Goldberg et al. , 2016 ). To maintain the OSs, photoreceptors need to ensure continuous transport of proteins and membranes from their site of synthesis in the IS through the CC to the OS ( Young, 1967 ). In addition to facilitating protein movement, the photoreceptor axoneme is thought to play a structural role in the formation and continual replacement of OS disks ( Liu et al. , 2004 ). Though the photoreceptor cilium is highly modified in function, the basic structure is consistent with immotile primary cilia seen in other tissues, containing 9 + 0 MT morphology that undergoes a switch from doublet microtubules (DMTs) to singlet MTs approximately one-third of the way up the axoneme ( Brown et al. , 1963 ; Steinberg and Wood, 1975 ; Knabe and Kuhn, 1997 ; Insinna et al. , 2008 ; Wensel et al. , 2016 ). The axonemal DMTs consist of an A-tubule containing 13 tubulin protofilaments joined to a B-tubule containing 10 tubulin protofilaments, with an 11th nontubulin complex linking the inner junction of A and B tubules (see Figure 7A later in this article) ( Linck and Stephens, 2007 ; Nicastro et al. , 2011 ; Pigino et al. , 2012 ; Linck et al. , 2014 ; Ichikawa et al. , 2017 ). Defects in the structural integrity of photoreceptor CC/axoneme lead to retinal degenerative diseases such as retinitis pigmentosa (RP), Lebers congenital amaurosis, and multiple ciliopathies ( Pierce et al. , 1999 ; Ramamurthy and Cayouette, 2009 ; Omori et al. , 2010 ; Boldt et al. , 2011 ; Bujakowska et al. , 2017 ). For example, mice lacking retinitis pigmentosa 1 (RP1), a protein that links the OS disks to the axoneme, displayed shortened axonemes and disordered OS disk structure ( Liu et al. , 2003 , 2004). Conversely, the absence of male germ-cell associated kinase (MAK) in murine retina resulted in extended axonemes, as well as disorganized OS disks ( Omori et al. , 2010 ). Despite the importance of the ciliary axoneme in photoreceptor structure and function, relatively little is known of the mechanism and players behind ciliogenesis and disk organization in the OS. Click here to read entire article References Anand M, Khanna H. (2012). Ciliary transition zone (TZ) proteins RPGR and CEP290: role in photoreceptor cilia and degenerative diseases. Expert Opin Ther Targets , 541–551. Bhamidipati A, Lewis SA, Cowan NJ. (2000). ADP ribosylation factor-like protein 2 (Arl2) regulates the interaction of tubulin-folding cofactor D with native tubulin. J Cell Biol , 1087–1096. Boldt K, Mans DA, Won J, van Reeuwijk J, Vogt A, Kinkl N, Letteboer SJ, Hicks WL, Hurd RE, Naggert JK, et al (2011). Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest , 2169–2180. Brown P, Gibbons I, Wald G. (1963). The visual cells and visual pigment of the mudpuppy, Necturus . J Cell Biol , 79–106. Bujakowska KM, Liu Q, Pierce EA. (2017). Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb Perspect Biol , a028274. Caspary T, Larkins CE, Anderson KV. (2007). The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell , 767–778. Datta P, Allamargot C, Hudson JS, Andersen EK, Bhattarai S, Drack AV, Sheffield VC, Seo S. (2015). Accumulation of non-outer segment proteins in the outer segment underlies photoreceptor degeneration in Bardet–Biedl syndrome. Proc Natl Acad Sci USA , E4400–E4409. Davidson AE, Schwarz N, Zelinger L, Stern-Schneider G, Shoemark A, Spitzbarth B, Gross M, Laxer U, Sosna J, Sergouniotis PI, et al (2013). Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet , 321–329. Dilan TL, Singh RK, Saravanan T, Moye A, Goldberg AFX, Stoilov P, Ramamurthy V. (2017). Bardet-Biedl Syndrome-8 (BBS8) protein is crucial for the development of outer segments in photoreceptor neurons. Hum Mol Genet , 283–294. Francis JW, Goswami D, Novick SJ, Pascal BD, Weikum ER, Ortlund EA, Griffin PR, Kahn RA. (2017). Nucleotide binding to ARL2 in the TBCD∙ARL2∙β-tubulin complex drives conformational changes in β-tubulin. J Mol Biol , 3696– 3716. Goldberg AFX, Moritz OL, Williams DS. (2016). Molecular basis for photoreceptor outer segment architecture. Prog Retin Eye Res , 52– 81. Hanke-Gogokhia C, Wu Z, Sharif A, Yazigi H, Frederick JM, Baehr W. (2017). The guanine nucleotide exchange factor, Arf-like protein 13b, is essential for assembly of the mouse photoreceptor transition zone and outer segment. J Biol Chem , 21442– 21456. Hong DH, Yue G, Adamian M, Li T. (2001). Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem , 12091–12099. Hsu Y, Garrison JE, Kim G, Schmitz AR, Searby CC, Zhang Q, Datta P, Nishimura DY, Seo S, Sheffield VC. (2017). BBSome function is required for both the morphogenesis and maintenance of the photoreceptor outer segment. PLoS Genet , e1007057. Ichikawa M, Liu D, Kastritis PL, Basu K, Hsu TC, Yang S, Bui KH. (2017). Subnanometre-resolution structure of the doublet microtubule reveals new classes of microtubule-associated proteins. Nat Commun , 15035. Insinna C, Pathak N, Perkins B, Drummond I, Besharse JC. (2008). The homodimeric kinesin, Kif17, is essential for vertebrate photoreceptor sensory outer segment development. Dev Biol , 160–170. Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, Cremers FP, Ropers HH, Berger W. (1999). RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet , 1571–1578. Knabe W, Kuhn H. (1997). Ciliogenesis in photoreceptor cells of the tree shrew retina. Anat Embryol (Berl) , 123–131. Ku CA, Chiodo VA, Boye SL, Hayes A, Goldberg AF, Hauswirth WW, Ramamurthy V. (2015). Viral-mediated vision rescue of a novel AIPL1 cone-rod dystrophy model. Hum Mol Genet , 670–684. Linck R, Fu X, Lin J, Ouch C, Schefter A, Steffen W, Warren P, Nicastro D. (2014). Insights into the structure and function of ciliary and flagellar doublet microtubules: tektins, Ca2+-binding proteins, and stable protofilaments. J Biol Chem , 17427–17444. Linck RW, Stephens RE. (2007). Functional protofilament numbering of ciliary, flagellar, and centriolar microtubules. Cell Motil Cytoskeleton , 489–495. Liu Q, Lyubarsky A, Skalet JH, Pugh EN, Pierce EA. (2003). RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci , 4171–4183. Liu Q, Zuo J, Pierce EA. (2004). The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci , 6427–6436. May-Simera H, Nagel-Wolfrum K, Wolfrum U. (2017). Cilia—the sensory antennae in the eye. Prog Retin Eye Res , 144–180. Meindl A, Dry K, Herrmann K, Manson E, Ciccodicola A, Edgar A, Carvalho MRS, Achatz H, Hellebrand H, Lennon A, et al (1996). A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet , 35. Molday RS, Moritz OL. (2015). Photoreceptors at a glance. J Cell Sci , 4039–4045. Nicastro D, Fu X, Heuser T, Tso A, Porter ME, Linck RW. (2011). Cryo-electron tomography reveals conserved features of doublet microtubules in flagella. Proc Natl Acad Sci USA , E845–E853. Nithianantham S, Le S, Seto E, Jia W, Leary J, Corbett KD, Moore JK, Al-Bassam J. (2015). Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble alphabeta-tubulin pool for microtubule dynamics. Elife , 1–33. Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T. (2010). Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci USA , 22671–22676. Pearring JN, Salinas RY, Baker SA, Arshavsky VY. (2013). Protein sorting, targeting and trafficking in photoreceptor cells. Prog Retin Eye Res , 24–51. Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. (1999). Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet , 248–254. Pigino G, Maheshwari A, Bui KH, Shingyoji C, Kamimura S, Ishikawa T. (2012). Comparative structural analysis of eukaryotic flagella and cilia from Chlamydomonas , Tetrahymena , and sea urchins. J Struct Biol , 199–206. Pinto LH, Invergo B, Shimomura K, Takahashi JS, Troy JB. (2007). Interpretation of the mouse electroretinogram. Doc Ophthalmol , 127–136. Prevo B, Scholey JM, Peterman EJG. (2017). Intraflagellar transport: mechanisms of motor action, cooperation, and cargo delivery. FEBS J , 2905–2931. Ramamurthy V, Cayouette M. (2009). Development and disease of the photoreceptor cilium. Clin Genet , 137–145. Rao KN, Zhang W, Li L, Ronquillo C, Baehr W, Khanna H. (2016). Ciliopathy-associated protein CEP290 modifies the severity of retinal degeneration due to loss of RPGR. Hum Mol Genet , 2005–2012. Sharer JD, Kahn RA. (1999). The ARF-like 2 (ARL2)-binding protein, BART. Purification, cloning, and initial characterization. J Biol Chem , 27553–27561. Steinberg R, Wood I. (1975). Clefts and microtubules of photoreceptor outer segments in the retina of the domestic cat. J Ultrastructural Res , 397–403. Stepanek L, Pigino G. (2016). Microtubule doublets are double-track railways for intraflagellar transport trains. Science , 721. Tian G, Thomas S, Cowan NJ. (2010). Effect of TBCD and its regulatory interactor Arl2 on tubulin and microtubule integrity. Cytoskeleton (Hoboken) , 706–714. Venkatesh A, Ma S, Langellotto F, Gao G, Punzo C. (2013). Retinal gene delivery by rAAV and DNA electroporation. Curr Protoc Microbiol Chapter 14, Unit-14D.4. Wensel TG, Zhang Z, Anastassov IA, Gilliam JC, He F, Schmid MF, Robichaux MA. (2016). Structural and molecular bases of rod photoreceptor morphogenesis and disease. Prog Retin Eye Res , 32–51. Wright ZC, Singh RK, Alpino R, Goldberg AFX, Sokolov M, Ramamurthy V. (2016). ARL3 regulates trafficking of prenylated phototransduction proteins to the rod outer segment. Hum Mol Genet , 2031–2044. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. (2013). One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell , 1370–1379. Young RW. (1967). The renewal of photoreceptor cells outer segments. J Cell Biol , 61–72. Zhang T, Li S, Zhang Y, Zhong C, Lai Z, Ding J. (2009). Crystal structure of the ARL2-GTP-BART complex reveals a novel recognition and binding mode of small GTPase with effector. Structure , 602–610. Zhao Y, Hong D-H, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T. (2003). The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA , 3965–3970. Zhou C, Cunningham L, Marcus AI, Li Y, Kahn RA. (2006). Arl2 and Arl3 regulate different microtubule-dependent processes. Mol Biol Cell , 2476–2487.
- A Novel Dominant Mutation in SAG, the Arrestin-1 Gene, Is a Common Cause of Retinitis Pigmentosa in Hispanic Families in the Southwestern United States
Lori S Sullivan, Sara J Bowne, Daniel C Koboldt, Elizabeth L Cadena, John R Heckenlively, Kari E Branham, Dianna H Wheaton, Kaylie D Jones, Richard S Ruiz, Mark E Pennesi, Paul Yang, David Davis-Boozer, Hope Northrup, Vsevold V Gurevich, Rui Chen, Mingchu Xu, Yumei Li, David G Birch, Stephen P Daiger | Invest Ophthalmology Vis Science | 2017 May 1 | Vol 58(5) | pgs. 2774-2784 | doi: 10.1167/iovs.16-21341 Abstract Purpose To identify the causes of autosomal dominant retinitis pigmentosa (adRP) in a cohort of families without mutations in known adRP genes and consequently to characterize a novel dominant-acting missense mutation in SAG . Methods Patients underwent ophthalmologic testing and were screened for mutations using targeted-capture and whole-exome next-generation sequencing. Confirmation and additional screening were done by Sanger sequencing. Haplotypes segregating with the mutation were determined using short tandem repeat and single nucleotide variant polymorphisms. Genealogies were established by interviews of family members. Results Eight families in a cohort of 300 adRP families, and four additional families, were found to have a novel heterozygous mutation in the SAG gene, c.440G>T; p.Cys147Phe. Patients exhibited symptoms of retinitis pigmentosa and none showed symptoms characteristic of Oguchi disease. All families are of Hispanic descent and most were ascertained in Texas or California. A single haplotype including the SAG mutation was identified in all families. The mutation dramatically alters a conserved amino acid, is extremely rare in global databases, and was not found in 4000+ exomes from Hispanic controls. Molecular modeling based on the crystal structure of bovine arrestin-1 predicts protein misfolding/instability. Conclusions This is the first dominant-acting mutation identified in SAG , a founder mutation possibly originating in Mexico several centuries ago. The phenotype is clearly adRP and is distinct from the previously reported phenotypes of recessive null mutations, that is, Oguchi disease and recessive RP. The mutation accounts for 3% of the 300 families in the adRP Cohort and 36% of Hispanic families in this cohort. Retinitis pigmentosa (RP) is the most common form of inherited retinal disease (IRD) and has a prevalence of approximately 1 in 4000 worldwide. 1 It is characterized by night blindness and progressive loss of peripheral vision, often leading to complete blindness. The presence of pigmentary deposits, attenuated retinal vessels, and changes to the ERG are typical clinical findings. 2 RP is exceptionally heterogeneous, with more than 100 genetic causes already described (RetNet: https://sph.uth.edu/retnet/ , in the public domain). Currently, mutations in 23 genes are known to cause dominant RP, 53 cause recessive RP, and 5 cause X-linked disease. At least 70 syndromic or systemic diseases include RP as a component. Variability in age of onset, secondary symptoms, rate of progression, and penetrance add to the complexity. Our autosomal dominant RP (adRP) cohort, described previously, 3 currently consists of 300 well-characterized families with evidence of an autosomal dominant form of disease. Likely pathogenic mutations have been identified in 226 of these families (75%). Of the 300 families, 195 (65%) have dominant mutations in known adRP genes, 25 (8%) have X-linked mutations, 3 (1%) have multiple segregating mutations, and 3 (1%) have dominant-acting mutations in genes previously associated with recessive diseases. By a combination of exome sequencing and targeted-capture next-generation sequencing (NGS) of known IRD genes we identified a novel dominant-acting mutation in the SAG gene, a gene previously thought to cause only recessive disease. 4 , 5 This mutation ( NM_000541.4 :c.440G>T; NP_000532.2 :p.Cys147Phe) appears to descend from a common ancestor and accounts for 8 of the 74 families in the cohort in which mutations had not been identified previously. Click here to read entire article References Daiger SP,, Bowne SJ,, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmology . 2007; 125: 151–158. Heckenlively JR. Retinitis Pigmentosa. Philadelphia, PA: J.B. Lippincott; 1988. Sullivan LS,, Bowne SJ,, Birch DG,, et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmology Vis Sci . 2006; 47: 3052–3064. Fuchs S,, Nakazawa M,, Maw M,, Tamai M,, Oguchi Y,, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet . 1995; 10: 360–362. Nakazawa M,, Wada Y,, Tamai M. Arrestin gene mutations in autosomal recessive retinitis pigmentosa. Arch Ophthalmology . 1998; 116: 498–501.
- Dissecting the role of EYS in retinal degeneration: clinical and molecular aspects and its implications for future therapy
Ana B. Garcia-Delgado , Lourdes Valdes-Sanchez , Maria Jose Morillo-Sanchez , Beatriz Ponte-Zuñiga , Francisco J. Diaz-Corrales , Berta de la Cerda | Orphanet Journal of Rare Diseases | 16, 222 | 17 May 2021 | https://doi.org/10.1186/s13023-021-01843-z Abstract Mutations in the EYS gene are one of the major causes of autosomal recessive retinitis pigmentosa. EYS-retinopathy presents a severe clinical phenotype, and patients currently have no therapeutic options. The progress in personalised medicine and gene and cell therapies hold promise for treating this degenerative disease. However, lack of understanding and incomplete comprehension of disease's mechanism and the role of EYS in the healthy retina are critical limitations for the translation of current technical advances into real therapeutic possibilities. This review recapitulates the present knowledge about EYS-retinopathies, their clinical presentations and proposed genotype–phenotype correlations. Molecular details of the gene and the protein, mainly based on animal model data, are analysed. The proposed cellular localisation and roles of this large multi-domain protein are detailed. Future therapeutic approaches for EYS-retinopathies are discussed. Background Retinitis pigmentosa (RP, OMIM #26800) is the most common form of inherited retinal degeneration (IRD), with an estimated prevalence of 1 per 4,000 people. Although RP is a rare disease, it represents the primary cause of hereditary blindness in adults, affecting more than one million people worldwide [ 1 ]. EYS is a major causative gene for autosomal recessive RP (arRP) [ 2 ] in all ethnicities. EYS-retinopathy manifests early in life and produces a severe disability, currently without therapeutic options. The study of the disease's molecular mechanism has been hampered by the lack of a representative animal model for this human IRD. Information on the cellular localisation and molecular features of EYS, obtained from different vertebrate animal models, is summarised in this review to get insight into this protein's possible roles in the human retina. Gene therapy is emerging as a safe and effective treatment for some types of RP caused by specific genes such as RPE65 . Future therapeutic approaches for EYS-retinopathies are discussed based on this large gene's limitations and the current advanced therapies state-of-the-art. Click here to read entire article References Menghini M, Cehajic-Kapetanovic J, MacLaren RE. Monitoring progression of retinitis pigmentosa: current recommendations and recent advances. Expert Opin Orphan Drugs. 2020;8(2–3):67–78. Abd El-Aziz MM, Barragan I, O’Driscoll CA, Goodstadt L, Prigmore E, Borrego S, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008;40(11):1285–7.
- New gene therapy technique
Shows promise in stem cell model of retinitis pigmentosa Although the 2017 FDA approval of Spark Therapeutics’ gene therapy Luxturna was considered a triumph for people with inherited eye disease, the product can only be used in patients with mutations in the RPE65 gene. Now, scientists in the United Kingdom say they’ve developed a gene therapy technique that could help a larger population of patients with retinitis pigmentosa. Scientists at Trinity College Dublin and University College London zeroed in on the RP2 gene, which can become mutated to cause a number of forms of retinitis pigmentosa. Functional copies of the RP2 gene delivered to “mini retinas” were able to pump out a vital protein called rhodopsin, the researchers reported (PDF) in the journal Stem Cell Reports. The mini retinas used in the study were three-dimensional organoids made from induced pluripotent stem cells and stem cells taken from patients with RP2-mutated retinitis pigmentosa. The organoids started losing the rod photoreceptor cells needed for proper vision at day 150. The gene therapy prevented that degeneration, the researchers said. To continue reading the article, click here .
- Genome Editing in Retinal Diseases using CRISPR Technology
Glenn Yiu, MD, PhD - Department of Ophthalmology & Vision Science, University of California, Davis, Sacramento, California | Ophthalmology Retina | Vol 2, 1 | January 2018 What is Genome Editing? Gene therapy has been the longstanding promise for not just treating, but providing a cure for retinal diseases. This is accomplished by using a delivery vector such as adeno- associated virus (AAV) to deliver a functional copy of a defective gene for inherited retinal degenerations, or to overexpress an antiangiogenic factor for neovascular con- ditions. One of the first ophthalmic applications of gene therapy was in patients with type 2 Leber congenital amaurosis, where subretinal delivery of an AAV expressing the RPE65 gene was successful in improving both sensi- tivity to light and functional vision. 1 Similar strategies have also been used to deliver the ABCA4 gene to patients with Stargardt disease, or the CHM gene in X-linked choroideremia. However, these approaches require continuous production of the protein product, and the duration of viral-mediated gene expression is uncertain. For example, long-term follow-up of Leber congenital amaurosis patients who received the RPE65 gene showed possible loss of efficacy after 2 to 3 years. 2 In contrast to traditional gene augmentation therapy, genome editing involves a direct modification to the DNA. Instead of overexpressing a functional protein to compen- sate for the mutated or defective gene product, genome editing using clustered regularly interspaced short palin- dromic repeat (CRISPR) systems repairs the actual disease- causing mutation in the genome. This technology is powerful for many reasons. Repairing mutations at the DNA level means that the repaired gene will be expressed and regulated physiologically, under the cell’s native pro- moter, rather than being overexpressed from an artificial promoter. The treatment effect is also theoretically per- manent, and the genetic modifications can propagate when the cell divides, even if the CRISPR system is no longer functional. Hence, genome editing represents a powerful and more refined gene therapy strategy with the potential to provide a true cure for retinal conditions. Click here to download the entire academic paper
- Progression of PROM1-Associated Retinal Degeneration as Determined by Spectral-Domain Optical Coherence Tomography Over a 24-Month Period
Manuel Großpötzl , Regina Riedl , Gernot Schließleder , Zhihong Jewel Hu , Michel Michaelides , SriniVas Sadda , David Birch , Peter Charbel Issa , Andreas Wedrich , Gerald Seidel a , Hendrik P.N. Scholl , Rupert W. Strauss | American Journal of Ophthalmology | Vol 259 | March 2024 | Pages 109-116 PURPOSE To evaluate the progression of atrophy as determined by spectral-domain optical coherence tomography (SD-OCT) in patients with molecularly confirmed PROM1 -associated retinal degeneration (RD) over a 24-month peri od. DESIGN International, multicenter, prospective case series. METHODS A total of 13 eyes (13 patients) affected with PROM1 -associated RD were enrolled at 5 sites and SD-OCT images were obtained at baseline and after 24 months. Loss of mean thickness (MT) and intact area were estimated after semi-automated segmentation for the following individual retinal layers in the central subfield (CS), inner ring, and outer ring of the ETDRS grid: retinal pigment epithelium (RPE), outer segments (OS), inner segments (IS), outer nuclear layer (ONL), inner retina (IR), and total retina (TR). RESULTS Statistically significant losses of thickness of RPE and TR were detected in the CS and inner ring and of ONL and IS in the outer ring (all P < .05); a statistically significant decrease in the intact area of RPE and IS was observed in the inner ring, and of ONL in the outer ring (all P < .05); the change in MT and the intact area of the other layers showed a trend of decline over an observational period of 24 months. CONCLUSIONS Significant thickness losses could be detected in outer retinal layers by SD-OCT over a 24-month period in patients with PROM1 -associated retinal degeneration. Loss of thickness and/or intact area of such layers may serve as potential endpoints for clinical trials that aim to slow down the disease progression of PROM1 -associated retinal degeneration. Click here to read entire article
- Precision Medicine:
Genetic Repair of Retinitis Pigmentosa in Patient-Derived Stem Cells Abstract Induced pluripotent stem cells (iPSCs) generated from patient fibroblasts could potentially be used as a source of autologous cells for transplantation in retinal disease. Patient-derived iPSCs, however, would still harbor disease-causing mutations. To generate healthy patient-derived cells, mutations might be repaired with new gene-editing technology based on the bacterial system of clustered regularly interspersed short palindromic repeats (CRISPR)/Cas9, thereby yielding grafts that require no patient immunosuppression. We tested whether CRISPR/Cas9 could be used in patient-specific iPSCs to precisely repair an RPGR point mutation that causes X-linked retinitis pigmentosa (XLRP). Fibroblasts cultured from a skin-punch biopsy of an XLRP patient were transduced to produce iPSCs carrying the patient’s c.3070G > T mutation. The iPSCs were transduced with CRISPR guide RNAs, Cas9 endonuclease and a donor homology template. Despite the gene’s repetitive and GC-rich sequences, 13% of RPGR gene copies showed mutation correction and conversion to the wild-type allele. This is the first report using CRISPR to correct a pathogenic mutation in iPSCs derived from a patient with photoreceptor degeneration. This important proof-of-concept finding supports the development of personalized iPSC-based transplantation therapies for retinal disease. Introduction Stem cell-derived cell transplantation in the eye is one therapy being explored for inherited retinal degenerations such as retinitis pigmentosa (RP). Recent clinical trials evaluating allogeneic retinal grafts derived from human embryonic stem cells (hESCs) show the procedure to be safe and potentially effective 1 . However, hESC-based treatments involve the controversial use of human embryos and pose a risk of immune-mediated rejection. Using a patient’s own cells for transplantation would avoid these pitfalls and is possible with induced pluripotent stem cells (iPSCs). Through established protocols 2 , fibroblasts from a skin biopsy can be returned to a pluripotent state and serve as a renewable, autologous source of replacement cells that avoids the ethical complications of hESCs. To continue reading this paper, click here .
- X-linked Retinitis Pigmentosa: RPGR Mutations in Most Families with Definite X Linkage and Clustering of Mutations in a Short Sequence Stretch of Exon ORF15
In grid Bader, Oliver Brandau, Helene Achatz, Eckart Apfelstedt-Sylla, Martin Hergersberg, Birgit Lorenz, Bernd Wissinger, Bärbel Wittwer, Günther Rudolph, Alfons Meindl, Thomas Meitinger | Invest Ophthalmology & Visual Science | 2003 Apr | Vol. 44 | 1458-1463 | doi.org/10.1167/iovs.02-0605 Purpose A comprehensive screening was conducted for RP2 and retinitis pigmentosa GTPase regulator (RPGR) gene mutations including RPGR exon ORF15 in 58 index patients. The frequency of RPGR mutations was assessed in families with definite X-linked recessive disease (xlRP), and a strategy for analyzing the highly repetitive mutational hot spot in exon ORF15 is provided. Methods Fifty-eight apparently unrelated index-patients were screened for mutations in all coding exons of the RP2 and the RPGR genes, including splice-sites, by single-strand conformation polymorphism (SSCP) analysis, except for RPGR exon ORF15. A strategy for directly sequencing the large repetitive stretch of exon ORF15 from a 1.6-kb PCR-product was developed. According to pedigree size and evidence for X linkage, families were subdivided into three categories. Results Screening of 58 xlRP families revealed RP2 mutations in 8% and RPGR mutations in 71% of families with definite X-linked inheritance. Mutations clustered within a ∼500-bp stretch in exon ORF15. In-frame sequence alterations in exon ORF15 ranged from the deletion of 36 bp to the insertion of 75 bp. Conclusions Mutations in the RPGR gene are estimated to cause 15% to 20% of all cases of RP, higher than any other single RP locus. This report provides a detailed strategy to analyze the mutational hot spot in RPGR exon ORF15, which cannot be screened by standard procedures. The discrepancy of the proportion of families linked to the RP3 locus and those having RPGR mutations is resolved in a subset of families with definite X linkage. Read more, click here
- First Gene Therapy FDA-Approved for an Inherited Retinal Disease
The approval has stimulated research into gene therapies for other IRDs. Meghan J. DeBenedictis, MS, LGC, MEd, and Aleksandra V. Rachitskaya, MD The idea of gene therapy has been discussed in the medical literature since as early as the 1970s. In 1972, Friedman and Roblin proposed that it was theoretically possible to introduce “good” DNA to replace defective DNA. Over the years, a number of gene therapy clinical trials emerged in efforts to treat genetic diseases of inborn errors of metabolism, all with varying degrees of success. The basic principle of gene therapy is to put corrective genetic material into cells to treat genetic disease. Several gene therapy approaches, including replacement gene therapy, optogenetics, addition of a growth factor, suppression gene therapy, and gene editing, have been proposed in attempts to treat various ophthalmologic conditions. Optogenetics focuses on creating artificial photoreceptors to restore photosensitivity. This is accomplished by gene delivery of light-activated optogenetic tools (channels or pumps) to surviving cells, such as ganglion cells, that remain intact in the retinal circuit in various diseases. It has been posited that genetically added growth factor proteins, such as adenovirus-expressed endostatin and angiostatin gene products, could have anti-VEGF properties. Short-interfering RNAs could be used for down-regulation of gene expression, resulting in functional inactivation of the targeted genes.4 Finally, technologies such as CRISPR (standing for clustered regularly interspaced short palindromic repeats ) could allow editing of one or several sites within the mammalian genome. Click here to read more entire article Meghan J. DeBenedictis, MS, LGC, MEd Licensed, Certified Genetic Counselor, Cole Eye Institute, Cleveland Clinic Foundation, Cleveland, Ohio marinom2@ccf.org Financial Disclosure: Consultant (Spark Therapeutics, Second Sight Medical Products) Aleksandra V. Rachitskaya, MD Vitreoretinal Physician, Cole Eye Institute, Cleveland Clinic Foundation, and Assistant Professor of Ophthalmology, Cleveland Clinic Lerner College of Medicine; both in Cleveland, Ohio rachita@ccf.org Financial Disclosure: None